



Personalisierte Medizin: Ein Strategiewechsel (Teil 3)
Molekulare Medizin: Aufnahme und Ausscheidung sind Schlüsselfaktoren für die individuelle Arzneimittel-wirksamkeit und Arzneimittelverträglichkeit
(Autor: Prof. Dr. Theo Dingermann)
Längst nicht jeder Wirkstoff, der im Reagenzglas und im biologischen Modell hervorragend funktioniert, wird auch beim Menschen „funktionieren“ oder wird von ihm vertragen. Und schon gar nicht lässt sich ein Wirk- und Verträglichkeitsprofil auf alle Menschen und Patienten übertragen. Der Grund liegt darin, dass der Wirkstoff zunächst sein Ziel im Organismus auf wesentlich komplizierterem Wege finden muss, um wirken zu können, als das im Reagenzglas der Fall ist. Zusätzlich muss er nach einer gewissen Zeit für die Ausscheidung vorbereitet werden, um ein möglichst gutes Verträglichkeitsprofil zu entfalten.
Verantwortlich für diese Anforderungen an einen guten Arzneistoff sind Proteine, die zum einen als Transporter fungieren, um entweder den Wirkstoff in die relevanten Zellen zu „holen“ oder ihn aus der Zelle auszuschleusen. Zum anderen spielen hier Proteine eine wichtige Rolle, die als Enzyme fungieren und chemische Reaktionen an den Wirkstoffen katalysieren, die die Eigenschaften dieser Moleküle so verändern, dass sie nun vom Organismus über die Nieren oder den Faeces ausgeschieden werden können.
Somit reicht es nicht aus, bei der Bewertung eines Arzneimittels nur auf die Interaktion des Wirkstoffs mit der therapierelevanten biologischen Zielstruktur zu schauen (pharmakodynamische Interaktion). Genauso wichtig ist es, auch die Interaktionen des Arzneimittels mit solchen Biomolekülen zu betrachten, die es dem Wirkstoff ermöglichen, die therapierelevante biologische Zielstruktur zunächst zu erreichen, und auf die Interaktion mit den Enzymen zu schauen, die den Wirkstoff chemisch modifizieren müssen, damit er den Organismus auch wieder verlassen kann (pharmakokinetische Interaktion).
Die hier relevanten Transporter und Enzyme sind durchaus sonderbar, da sie im Vergleich zu anderen Biomolekülen hinsichtlich ihrer Substratwahl meist relativ unspezifisch sind und nicht selten eine Vielzahl sehr unterschiedlicher Substrate erkennen. Daher zeigen oft sehr viele Arzneimittel ein untypisches pharmakokinetisch / pharmakodynamisches Verhalten mit teils signifikanten Konsequenzen für die Wirksamkeit und Verträglichkeit, wenn diese Proteine bei einem bestimmten Patienten aufgrund einer Mutation nicht wie die „Norm“ funktionieren.
In dieser Folge wollen wir uns ein wenig genauer mit den Transportern auseinandersetzen und an zwei Beispielen erläutern, wie diese Moleküle ein Wirkstoffprofil verändern können, wenn sie nicht richtig funktionieren. In der Folge 4 dieser Serien sollen dann die Enzyme des Arzneimittelmetabolismus genauer betrachtet werden.
Transporter können Wirkstoffe in die Zelle hineinholen oder sie aus einer Zelle ausschleusen
Im Laufe der Evolution haben sich Proteine entwickelt, die dafür sorgen, dass giftige Substanzen, die in eine Zelle gelangt sind, schnell wieder ausgeschleust werden. Es haben sich aber auch Proteine entwickelt, die bestimmte Stoffe förmlich in die Zellen hineinsaugen. Beide Transporter werden auch von Arzneimitteln benutzt, so dass es zu Unregelmäßigkeiten kommen kann, wenn ein Patient ein solches Protein in einer funktionsunfähigen Form synthetisiert, oder wenn es gar nicht synthetisiert wird.
Transporter, die Wirkstoffe aus einer Zelle ausschleusen
Der wohl bekannteste Vertreter der Gruppe von Transportern, die Fremdmoleküle aus eine Zelle ausschleusen, ist das „Multidrug-Resistenzprotein“, das auch als „p-Glykoprotein“ bezeichnet wird. Dieses p-Glykoprotein ist ein Vertreter der so genannten ABC-Protein-Familie (ATP-binding-cassette-proteins). Diese Proteine transportieren ihre Substrate unter ATP-Verbrauch (Abb. 1).
Entdeckt wurde das p-Glykoprotein als Grund für die oft beobachtete Resistenz von Zytostatika bei der Tumortherapie. Daher stammt auch der Name Multidrug-Resistenzprotein (MDR). Jedoch wird dieses Protein keineswegs nur in der Membran von Tumorzellen gefunden. Es kommt in großer Anzahl in Membranen von Zellen vor, die eine exkretorische (ausscheidende) oder schützende Funktion haben, so in der Niere, in Darmepithelzellen, in der Gallenkapillarmembran von Leberzellen, in der Plazenta oder in den Zellen der Blut-Hirn-Schranke.
Das p-Glykoprotein bildet damit einen wesentlichen Eliminationsfaktor in unserem Körper und ist somit auch extrem relevant für die Bioverfügbarkeit von Arzneimitteln. Zu den Medikamenten, die von p-Glykoprotein eliminiert werden, gehören Opioide, Steroide, Antibiotika, Kalziumkanal-Blocker, Zytostatika, Immunsystem hemmende Substanzen, Medikamente gegen das Aids verursachende HI-Virus, Beta-Blocker und viele andere mehr (siehe Tabelle 1). Sollten Mutationen vorliegen, die dieses Protein in seiner Aktivität herabsetzen, kann mit einer gesteigerten Bioverfügbarkeit gerechnet werden, da ja der Auswärtstransport aus der Zelle beeinträchtigt ist. Allerdings wird eine Beurteilung der Konsequenzen einer detektierten Mutation dadurch erschwert, dass es eine ganze Reihe weiterer ABC-Transporter gibt, die die Funktion des mutierten Proteins teilweise oder ganz übernehmen können. Detektierte Mutationen können daher bestenfalls auf ein mögliches Problem hinweisen. In einem solchen Fall sollte die Therapie hinsichtlich Wirksamkeit und Verträglichkeit eines p-Glykoprotein-Substrates aufmerksam beobachtet und bei Auffälligkeiten die Dosis gegebenenfalls angepasst werden.
Transporter, die Wirkstoffe in eine Zelle hineintransportieren
Gegenteilig zur Funktion der ABC-Transporter verhalten sich beispielsweise so genannte „Organische Anionen-Transporter“, die bei Aufnahme von Statinen in die Leber eine Rolle spielen.
Zwar wirken alle Statine pharmakodynamisch gleich, indem sie die HMGCoA-Reduktase, das Schlüsselenzym bei der Cholesterinbiosynthese, hemmen. Allerdings unterscheiden sie sich teilweise deutlich hinsichtlich ihrer Pharmakokinetik.
An dem pharmakokinetischen Verhalten der verschiedenen Statine sind organische Anionen-Transporter maßgeblich beteiligt. Sie sind dafür verantwortlich, dass sich Statine selektiv in der Leber anreichern – zumindest dann, wenn die Transportproteine in einer funktionstüchtigen Form vorliegen. Ein gut untersuchter Vertreter dieser Transporter ist OATP2 (organic anion transporter 2), den man in der Literatur auch unter dem Namen SLCO1B1 (solute carrier organic anion transporter family member 1B1) findet. Zu diesem Transporter besitzt das Statin Simvastation, das unter physiologischen Bedingungen vorwiegend als Carboxylat-Anionen vorliegt, eine hohe Affinität. Auch für Pitavastatin wird ein solcher Mechanismus angenommen, da die gleichzeitige Applikation von Ciclosporin, ein OATP2-Inhibitor, zu einem mehr als 4-fachen Anstieg der Plasmakonzentration des Statins führt. Aus diesem Grund ist Pitavastatin bei Patienten kontraindiziert, die mit Ciclosporin behandelt werden (Hirano et al. 2004).
Ist das Gen für den SLCO1B1-Transporter nur in einer Kopie oder gar nicht in einer funktionsfähigen Form vorhanden, kommt es ebenfalls zu deutlich erhöhten peripheren Statin-Konzentrationen. Daraus resultiert ein relevantes Risiko für Myopathien bis hin zu Rhabdomyelosen, die als lebensbedrohliche Komplikationen einzustufen sind. (Abb.2).
Zusätzlich sind einige Statine Substrate für Auswärtstransporter des bereits besprochenen ABC-Transportertyps. Tragen auch diese Auswärtstransporter Mutationen, verschärft sich die Nebenwirkungsproblematik. Unter „normalen“ Bedingungen limitieren die ABC-Transporter die Bioverfügbarkeit der Statine und erhöhen die renale Sekretion dieser Wirkstoffe. Liegen Mutationen vor, steigen die peripheren Statin-Konzentrationen. So wurden beispielsweise bis zu ca. 145 % erhöhte AUC-Konzentrationen von Rosuvastatin und bis zu ca. 110 % erhöhte AUC-Konzentrationen von Simvastation-Lacton bei Vorliegen eines c.421AA-Genotyps des ABCG2-Transporters gemessen.
Ganz ähnlich erhöhen sich die systemischen Statin-Konzentrationen bei Vorliegen von ABCA1-Mutationen, da auch dieser Transporter Statine als Substrate bindet.
Aus diesen Überlegungen schlägt M. Niemi in der Clinical Pharmacology & Therapeutics erschienenen Publikation „Transporter Pharmacogenetics and Statin Toxicity“ für Kaukasier folgende Dosisanpassungen (Maximaldosis) beim Vorliegen eines SLCO1B1 c.521T>C Genotyps vor:
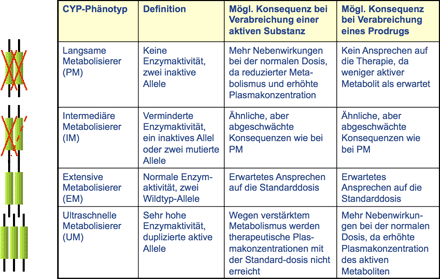
Das Beispiel der unterschiedlichen pharmakokinetischen Eigenschaften der verschiedenen Statine macht auch deutlich, wie vorsichtig man mit dem Begriff „Me-too-Arzneimittel“ umgehen sollte. Obwohl alle Statine qualitativ pharmakodynamisch analog wirken (darauf bezieht sich der Begriff „Me-too“), verhalten sie sich pharmakokinetisch dramatisch unterschiedlich. Dieses unterschiedliche Verhalten kann man nutzen, wenn sich aufgrund genetischer Charakteristika Probleme bei der Einnahme eines speziellen Statins ergeben sollten. Man muss dann nicht zwingend die Wirkstoffklasse verlassen, um das Problem zu umgehen, sondern kann bei Kenntnis der pharmakokinetischen Charakteristika auf einen anderen Wirkstoff innerhalb der gleichen Klasse – gegebenenfalls nach Dosisanpassung – wechseln.
Hirano, M. et al. J Pharmacol Exp Ther 2004, 311, 139
M Niemi, Clinical Pharmacology & Therapeutics 2010, 87, 130
Abbildung 1
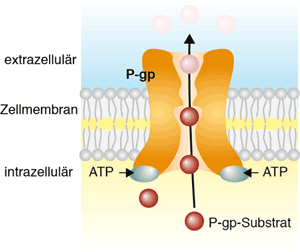
Abb. 1 Schematische Darstellung einen ABC-Transporters. Diese Proteine transportieren unter ATP-Hydrolyse Substarte, die über eine Diffusion durch die Zellmembran in die Zelle gelangt sind, wieder aus der Zelle hinaus.
Tabelle 1
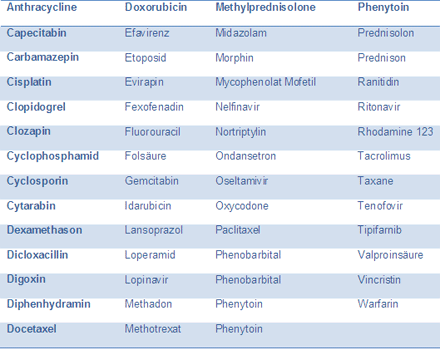
Tabelle 1 Substrate des p-Glykoproteins
Abbildung 2
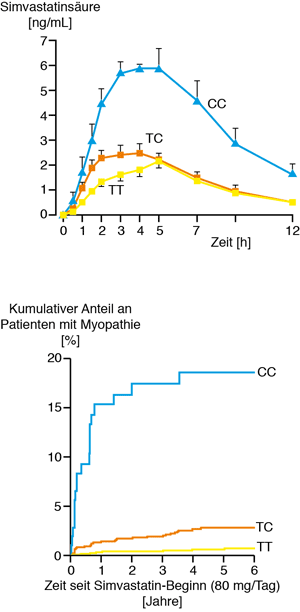
Abb. 2 Schematische Darstellung des SLCO1B1-Transporters und die peripheren Simvastatin-Konzentrationen sowie das Auftreten von Myopathien in Abhängigkeit vom Genotyp.
Abbildung 3
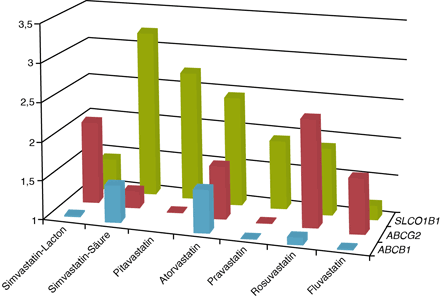
Abb. 3 Effekte von Mutationen in dem organischen Anionentransporter SLCO1B1 und in den Eflux-Transportern ABCG2, und ABCB1 auf die systemische Konzentration verschiedener Statine . Dargestellt ist jeweils das Vielfache der AUCs im Vergleich zur Wildtyp-Situation Vorliegen einer SLCO1B1-c.521CC-Mutation (grün), einer ABCG2-c.421AA-Mutation (rot) und eines ABCB1-c.1236TT-c.2677TT-c.3435TT-Haplotyps. Die entsprechenden Wildtypgenotypen sind SLCO1B1-c.521TT, ABCG2-c.421CC sowie ABC1B1-c.1236CC-c.2677GG-c.3435CC.
