



Personalisierte Medizin: Ein Strategiewechsel (Teil 4)
Molekulare Medizin: Metabolismus
(Autor: Prof. Dr. Theo Dingermann)
Gelangt ein Wirkstoff in den Organismus, wird dieser bemüht sein, dieses fremde Molekül möglichst schnell wieder zu eliminieren. Dies ist das Ergebnis einer langen evolutionären Entwicklung, denn die Natur hält ein riesiges Arsenal an gefährlichen Molekülen bereit, die auch in Quellen vorkommen, von denen sich der Mensch ernährt. Verantwortlich für die Eliminationsan-strengungen sind die Enzyme des metabolisierenden Systems, die sich in zwei Gruppen aufteilen lassen: Die Enzyme des Phase I-Metabolismus und die Enzyme des Phase II-Metabolismus.
Phase-I-Enzyme, zu denen vor allem die Enzyme der Cytochrom P450-Familie gehören, modifizieren Moleküle durch Einführung funktioneller Gruppen (meist durch Oxidation).
Diese funktionellen Gruppen werden von Phase-II-Enzymen genutzt, um daran hydrophile Moleküle (z. B. Glucuronsäure) anzuknüpfen. Dieser Prozess wird auch als Konjugation bezeichnet. Auf diese Weise werden hydrophile und biologisch inaktive Konjugate erzeugt, die renal oder biliär eliminiert werden können.
In beiden Metabolisierungsgruppen gibt es Vertreter, die genetisch polymorph sind, so dass die Enzymausstattung in der Bevölkerung alles andere als einheitlich ist (Abb. 1). Daraus folgert, dass auch die mittlere Verweilzeit eines Wirkstoffs im Körper eines Patienten uneinheitlich ist, woraus gravierende Probleme hinsichtlich Wirksamkeit und Verträglichkeit erwachsen können.
Abbildung 1: Phase l
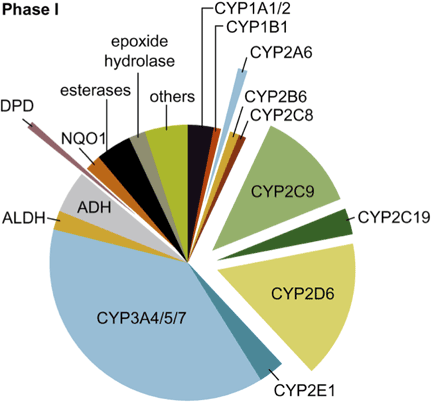
Abbildung 1: Phase ll
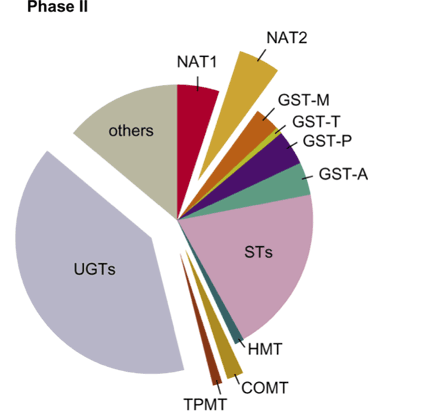
Abb 1 Die wichtigsten Vertreter der metabolischen Phase I- und Phase II-Enzyme. Neben den Vertretern des Cytochrom-P450-Familie findet man unter den Phase-I-Enzymen die Alkohol-Dehydrogenase (ALDH), die Aldehyd-Dehydrogenase (ADH), die Dihydropyrimidin-Dehydrogenase (DPD, die NAD(P)H-Dehydrogenase , quinone 1 (NQO1), verschiedene Esterasen, Epoxid-Hydrolasen und andere Enzyme.
Unter den Phase-II-Enzymen findet man die UDP-Glucuronosyltransferasen (UGTs), die N-Acetyltransferasen (NAT1 und NAT2), die Glutathiontransferasen (GST-M, -T, -P, -A), die Sulfotransferasen (STs), die Histamin-Methyltransferase (HAT), die Catechol-O-Methyltransferase (COMT), die Thiopurin-Methyltransferase (TPMT) und andere Enzyme.
Diejenigen Enzyme, die einen relevanten genetischen Polymorphismus aufweisen, ragen aus dem Tortendiagramm heraus.
Ist beispielsweise ein metabolisierendes Enzym bei einem Patienten aufgrund des Vorliegens einer inaktivierenden Mutation unterrepräsentiert, wird der Wirkstoff viel langsamer ausgeschieden, als das normalerweise der Fall ist. Dosiert der Patient in einem solchen Fall seine Medikation nach Vorschrift, wird es bei diesem Patienten langsam zu einer teils gefährlichen Akkumulation des Wirkstoffs kommen, was u.a. in Form von unerwünschten Arzneimittel-wirkungen wahrgenommen wird.
Da die Vertreter der Cytochrom P450-Familie auch an der Aktivierung eines Prodrugs beteiligt sind, kann es bei Vorliegens einer inaktivierenden Mutation in einem relevanten Cytochrom P450-Enzym zu Defiziten kommen, die letztlich eine Non-Response nach sich ziehen. In einem solchen Fall wird der Patient letztlich nicht behandelt, obwohl er ein Medikament eingenommen hat.
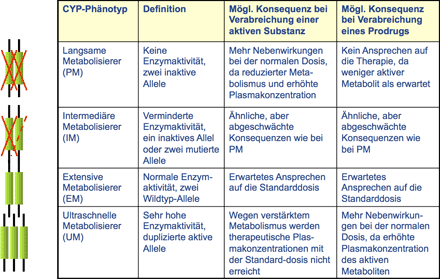
Man unterscheidet vier verschiedene Phänotypen, je nach dem wie viele Genkopien für ein bestimmtes metabolisierendes Enzym vorliegen:
- Sind zwei aktive Kopien im Genom des Patienten vorhanden, spricht man von einem Extensiven-Metabolisierer-Phänotyp (EM).
- Bei Vorliegen nur einer aktiven Kopie im Genom des Patienten, spricht man von einem Intermediären-Metabolisierer-Phänotyp (IM).
- Bei Vorliegen nur keiner aktiven Kopie im Genom des Patienten, spricht man von einem Langsamen-Metabolisierer-Phänotyp (PM).
- Liegen nach Genduplikationen mehr als zwei aktive Kopien im Genom des Patienten vor, spricht man von einem Ultraschnellen-Metabolisierer-Phänotyp (UM).
Die Konsequenzen dieser Konstellationen sind in Abb. 2 zusammengefasst:
Abbildung 2
Der promiskuide Charakter der metabolisierenden Enzyme
Viele der metabolisierenden Enzyme sind bemerkenswert in der Hinsicht, dass sie eine ungewöhnlich große Zahl an Substraten erkennen. So metabolisiert das Enzym CYP3A4 nahezu 50 % der zugelassen Wirkstoffe, das Enzym CYP2D6 25 – 30 % der zugelassenen Wirkstoffe und das Enzym CYP2C9 ca. 15 % der zugelassenen Wirkstoffe. Daraus leitet sich ab, dass man bei Kenntnis des Metabolisierer-Phenotyps für ein bestimmtes Enzym Aussagen zum pharmakokinetischen Verhalten für eine Vielzahl von Wirkstoffen treffen kann.
Eine gute Zusammenstellung der wichtigsten Substrate für die wichtigsten Cytochrom-P450-Enzyme findet man hier: http://medicine.iupui.edu/clinpharm/ddis/table.aspx. Kennt man seinen Genotyp hinsichtlich metabolisierender Enzyme und nimmt diese Tabelle zur Hand, kann man sehr leicht ableiten, bei welchen Medikamenten es zu Unregelmäßigkeiten hinsichtlich der Pharmakokinetik dieser Wirkstoffe kommen kann, und welche Maßnahmen in Betracht gezogen werden können, um diese Probleme zu minimieren.
Beispiel: CYP2D6-Defizienz und Einsatz von Codein zur Schmerztherapie
In den USA mehr als in Europa wird Codein zur Schmerztherapie eingesetzt. Dies beruht darauf, dass Codein als Prodrug von CYP2D6 zu Morphin metabolisiert wird. Somit ist Morphin verantwortlich für die analgesierende Wirkung des Codeins.
Daraus folgt, dass langsame Metabolisierer (PMs) Codein nicht zu Morphin umwandeln können und somit Non-Responder für Codein sind. Heute wird dies empirisch ermittelt, was für die Betroffenen fürchterlich sein kann, da sie trotz Therapie keine Analgesie verspüren.
Abbildung 3
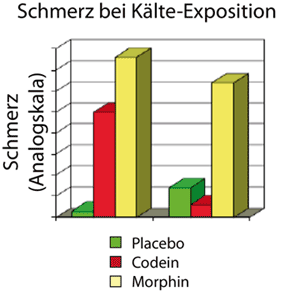
Abb. 3 Die Scherzintensität nach Kälte-Exposition wurde mit Hilfe einer Analogskala ermittelt. Je höher der Balken, um so geringer ist der Schmerz. Der linke Balkenblock wurde an Extensiven Metabolisierern (EM) ermittelt, wohingegen der rechte Balkenblock an langesamen Metabolisierern ermittelt wurde. Man sieht deutlich, dass bei den langsamen Metabolisiern Codein fast keine analgesierende Wirkung entfaltet, da das Prodrug nicht zu Morphin metabolisiert werden kann.
Ein analoges Beispiel findet sich im zweiten Teil dieser Serie im Falle von Tamoxifen.
Beispiel: CYP2D6-Defizienz und Einsatz von Doxepin bei der Behandlung von Angst- und Depressionszuständen.
Doxepin ist ein Hemmstoff der Monoamin-Wiederaufnahme aus dem synaptischen Spalt in die präsynaptischen Vesikel. Durch die unselektive Hemmung der Wiederaufnahme erhöht sich die Konzentration von Serotonin und Noradrenalin im synaptischen Spalt und führt zu einer Milderung depressiver Symptome.
Doxepin wird maßgeblich von CYP2D6 metabolisiert. Liegt dieses Enzym – wie bei langsamen Metabolisierern der Fall – nicht in „normaler“ Konzentration vor, so wird der Metabolismus stark verlangsamt und die Substanz entsprechend langsamer eliminiert. Nimmt ein betroffener Patient in Unkenntnis dieser Zusammenhänge das Medikament nach Vorschrift ein, erhöhen sich die systemischen Wirkstoffkonzentrationen erheblich, was in teils schweren unerwünschten Arzneimittelwirkungen resultiert. Die therapeutischen Plasma-Konzentrationen von Doxepin + Nordoxepin (aktiver Metabolit) liegen bei 50-150 ng/mL. Toxische Konzentrationen liegen vor, wenn die Konzentrationen von Doxepin + Nordoxepin Werte von größer oder gleich 500 ng/mL erreichen.
Die Dutch Pharmacogenetics Working Group hat aus diesem Problem mittlerweile Konsequenzen gezogen und gibt folgende Empfehlungen:
Tabelle 1
| Phenotyp (Genotyp) | Empfohlene Maßnahmen |
| PM (zwei inaktive (*3-*8, *11-*16, *19-*21, *38, *40, *42) Allele) | Dosisreduktion um 60%. Anpassung der Erhaltungsdosis entsprechend der Doxepin und Nordoxepin Plasma-Konzentrationen. |
| IM (zwei minderaktive Allele (*9, *10, *17, *29, *36, *41) oder ein aktives (*1, *2, *33, *35) und ein inaktives (*3-*8, *11-*16, *19-*21, *38, *40, *42) Allel, oder ein minderaktives Allel (*9, *10, *17, *29, *36, *41) und ein inaktives Allel (*3-*8, *11-*16, *19-*21, *38, *40, *42)). | Dosisreduktion um 20%. Anpassung der Erhaltungsdosis entsprechend der Doxepin und Nordoxepin Plasma-Konzentrationen. |
| UM (eine Genduplikation in Abwesenheit von inaktiven Allelen (*3-*8, *11-*16, *19-*21, *38, *40, *42) oder minderaktiven Allelen (*9, *10, *17, *29, *36, *41)). | Substitution von Doxepin durch einen alternativen Wirkstoff (Citalopram, Sertralin) oder Erhöhung der Dosis um 100%. Anpassung der Erhaltungsdosis entsprechend der Doxepin und Nordoxepin Plasma-Konzentrationen. |
Beispiel: Dihydropyrimidin-Dehydrogenase-Defizienz und Einsatz von 5-Fluorourazil im Rahmen einer Tumortherapie.
Bei ca. 3 – 5 % aller mit 5-Fluoruracil (5-FU, 5FU) therapierten Patienten werden toxische Reaktionen (Mukositis, neurologische Störungen, Kardiotoxizität), begleitet von einer Uracil- und Thyminurie beobachtet. Der Grund dieser unerwünschten Arzneimittelwirkungen ist eine erniedrigte Aktivität des Enzyms Dihydropyrimidin-Dehydrogenase (DPD). Dies resultiert in einem verlangsamten physiologischen Abbau des 5-FU (langsamer Metabolisierer-Phänotyp). Bei Vorliegen zweier aktiver DPD-Kopien werden in der Regel mehr als 80 % des verabreichten 5-FUs in kurzer Zeit metabolisiert. Patienten, die aufgrund inaktivierender Genmutationen eine zu niedrige DPD Aktivität aufweisen, akkumulieren das 5-FU im Plasma stark, was ernste Komplikationen nach sich ziehen kann.
Auch diesem Problem hat sich die Dutch Pharmacogenetics Working Group gewidmet und gibt folgende Empfehlungen
Tabelle 2
| Phenotyp (Genotyp) | Empfohlene Maßnahmen |
| PM (zwei inaktive Allele oder zwei minderaktive Allele oder ein inaktives Allel und ein minderaktive Allel) | Wahl eines alternativen Wirkstoffs, nicht jedoch Tegafur, der analog metaboliert wird. |
| IM (ein aktives Allel und ein minderaktive oder inaktives Allele) | Dosisreduktion um 50% oder Wahl eines alternativen Wirkstoffs, nicht jedoch Tegafur, der analog metaboliert wird. Danach Dosiserhöhung unter Beobachtung von Toxizität und Effektivität. |
Aktive Allele sind die Allele *1, *4, *5, *6, *9A
Minderaktive Allele sind die Allele *9B, *10
Inaktive Allele sind die Allele *2A, *3, *7, *8, *11, *12, *13, 496A>G, IVS10-15T>C, 1156G>T, 1845G>T
*http://www.pharmgkb.org/home/dutch_pharmacogenetics_working_group.jsp
